DEFINITIE
Malformatiile sistemului nervos central sunt definite ca anomalii morfologice legate de oprirea in dezvoltare sau dezvoltarea anormala a structurilor cerebrale cu aspect clinic heterogen si cauze variate. Acestea trebuie diferentiate de distrugerile structurilor deja formate care, in mod normal, n-ar trebui incluse in modulul malformatiilor (de exemplu o porencefalie cauzata de o ischemie, urmata apoi de o resorbtie a parenchimului cerebral, nu este o malformatie adevarata). Acesta distinctie este formala din doua motive:
1.
2. nu este intotdeauna usor de diferentiat o tulburare de geneza (malformatie) de una de distrugere ulterioara genezei, deoarece in acest ultim caz se produce resorbtie tisulara fara cicatrice gliala.
In practica este preferabil de a grupa sub termenul de malformatii toate anomaliile morfologice ale creierului constituite inainte de nastere.
Ar fi ideala o clasificare etiologica dar care in prezent nu este posibila. De aceea, cele mai multe clasificari apeleaza la embriologie, impartind malformatiile in doua grupe, prima, cuprinzand malformatii care se produc in primele 20 de saptamani de gestatie si a doua, cuprinzand malformatii produse in ultimele 20 de saptamani ale sarcinii.
In primele 20 de saptamani de gestatie se succed etapele de:
- neurulatie si formare a tubului neural;
- formarea culelor cerebrale;
- formarea si divizarea telencefalului in doua emisfere;
- diferentierea comisurilor din placa comisurala;
Perturbarea acestor etape duce la malformatii morfologice majore. Aceasta prima jumatate a sarcinii este de asemenea aceea de neurogeneza (multiplicarea neuroblastilor din zona germinativa periventriculara) si de migrare a lor de-a lungul ghizilor gliali pentru a constitui scoarta cerebrala. Acestea sunt malformatiile adevarate (precoce) ale SNC.
In ultimele 20 de saptamani se produc asa-numitele malformatii tardive prin distrugeri cerebrale focale si resorbtia tisulara la nivelul sistemului nervos deja format cu formarea unor cavitati chistice (a se vedea modulul Encefalopatia hipoxic-ischemica perinatala).
MALFORMATII PRECOCE
I. Tulburari ale neurulatiei si formarii tubului neural
Cuprind defecte de inchidere completa a tubului neural cu dezvoltare anormala a structurilor SNC. In absenta inchiderii tubului neural structurile mezenchimale nu se dezvolta corespunzator, astfel incat osul nu acopera neuroectodermul.
Termenul de disrafie implica o continuitate intre neuroectoderm si ectodermul cutanat. Disrafiile sunt: craniene (anencefalia si cefalocelel 515b14f e) si spinale (spina bifida chistica, oculta cu subtipurile sale). Cauzele disrafiilor sunt insuficient cunoscute, dar este cert ca factorii genetici au un rol important, modalitatea de transmitere fiind in general poligenica. Un procent mic de cazuri recunosc o transmitere mendeliana recesiva sau chiar X-linkata. Unele cazuri au mod de transmitere multifactorial, printre factorii de mediu incriminati fiind: varsta mica a mamei, nivelul socio-economic scazut, deficienta in vitamine, in special in acid folic, factori chimici si fizici.
Diagnosticul prenatal a defectelor inchiderii tubului neural este posibil prin ecografie fetala si determinarea alfa-fetoproteinelor (AFP) din lichidul amniotic obtinut prin amniocenteza. AFP reprezinta 90% din totalul globulinelor serice ale fatului. In cazul defectelor de tub neural, AFP trec in lichidul amniotic si apoi in sangele matern. Determinarea AFP in sangele matern este un test screening pentru depistarea defectelor de tub neural, niveluri sanguine peste 1000 ng/ml fiind considerate patologice, moment in care se indica efectuarea amniocentezei, care pune diagnosticul in 99% din cazuri daca determinarea este efectuata intre 16-l8 saptamani de varsta gestationala.
I.1. Disrafii craniene
Anencefalia rezulta din defectul de inchidere a tubului neural in portiunea sa craniala. Ca urmare celulele nervoase degenereaza, iar tesutul mezodermal lipseste, nemaifiind posibila formarea osului. In cazurile tipice de anencefalie craniul lipseste, iar emisferele cerebrale sunt inlocuite de un tesut conjunctiv rosietic hipervascularizat in care se gasesc resturi meningeale si neuronale. De obicei raman resturi de diencefal, puntea, bulbul, cerebelul si maduva spinarii. Frecvent se asociaza tulburari de inchidere ale vertebrelor si hernieri ale tesutului cerebral.
Aspectul clinic consta in absenta neurocraniului, dar cu dezvoltarea normala a viscerocraniului. Reflexele arhaice sunt prezente (reflexul de supt, reflexul Moro). Se pot evidentia crize cu aspect mioclonic. Anencefalii traiesc intre cateva ore pana la cateva saptamani.
Cefalocel (encefalocel, cranium bifidum) este un disrafism in care exista hernierea durei, creierului sau cerebelului in afara cutiei craniene (ura: Meningoencefalocel occipital).

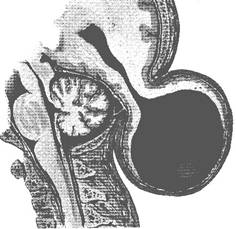
ura: Meningoencefalocel occipital
Se asociaza frecvent cu alte malformatii ca: agenezia corpului calos sau anomalii de giratie, sindrom Dandy-Walker, Arnold Chiari, disrafii de linie mediana. Cefalocelele pot avea diferite localizari. Localizarea occipitala poate fi supra- sau subtentoriala. Este mai frecvent in tarile vestice. Dimensiunea variaza de la cativa milimetri la mai mult de 10 centimetri. Pentru aprecierea continutului cefalocelului se foloseste transiluminarea si CT cerebral. Radiografia de craniu evidentiaza dehiscenta osoasa. Localizarea sfenoidala este rara, fiind evidentiata prin obstructia nazala, hipertelorism. Continutul este pulsatil si creste in volum in timpul sului. Se asociaza frecvent colobomul, hipoplazie oculara sau orbitara uni- sau bilaterala sau chiar o insuficienta hipofizara. Radiografia craniana si CT arata dehiscenta seului seii turcesti in incidenta din fata si o masa faringiana in incidenta de profil. Cazurile fistulizate cu rinoree si risc de meningita constituie indicatie chirurgicala. In localizarea fronto-etmoidala (observate in special in extremul orient) hernierea poate realiza un pseudopolip nazal descoperit la nastere si nasterea la domiciliu" class="text">nastere si nasterea la domiciliu" class="text">nastere si nasterea la domiciliu" class="text">nastere sau o masa voluminoasa frontala situata la nivelul suturii metopice. Localizarile parietala si bazala sunt rare.
In functie de continutul pungii herniare, cefalocelele sunt: encefalocele (contin tesut cerebral) si meningocele craniene (contin numai meninge si LCR, fara tesut cerebral). Acestea din urma sunt mai rare decat encefalocelele.
Tratamentul cefalocelului este chirurgical, rezultatele depinzand de volumul tesutului cerebral herniat si de malformatiile asociate.
I.2. Disrafii spinale
Termenul de disrafii spinale se aplica unui grup heterogen de anomalii spinale care au ca trasatura comuna formarea imperfecta a structurilor liniei mediane, mezenchimale, osoase si neurale.
Spina bifida chistica este tipul cel mai frecvent al disrafismului spinal si cuprinde in functie de continutul sacului herniar: mieloschizis, mielomeningocel si meningocel .
Mielomeningocelul si mieloschizisul constituie 90% dintre cazurile de disrafism spinal. Ele au structura identica, diferenta intre ele constand in faptul ca mieloschizisul este , in timp ce mielomeningocelul bombeaza. Sacul herniar contine meninge, maduva si radacini nervoase (ura: mielomeningocel lombar), de obicei neacoperite de piele. Meningele sunt foarte subtiri, se pot rupe usor si se pot infecta. Pielea din jur este de obicei anormala. Defectul osos este intins.


ura: Mielomeningocel lombar
Meningocelul este o hernie a meningelui spinal impreuna cu LCR, fara nici un element nervos in interiorul sacului herniar, care este acoperit de piele. Cea mai frecventa localizare este posterioara lombara, cele anterioare sunt posibile, dar extrem de rare.
Tabloul clinic contine tulburari motorii si senzitive. Cele motorii constau cel mai adesea intr-o paraplegie flasca, cu abolirea ROT. Daca sacul herniar este sus situat, paraplegia poate fi spastica cu hiperreflectivitate osteotendinoasa. Membrele inferioare au pozitii vicioase: picior var-equin, talus-valg. Se asociaza tulburari senzitive, nivelul lor determinand nivelul atingerii medulare. Tulburarile sfincteriene sunt constante, cu aparitie precoce, dar dificil de apreciat la nou-nascut, la care exista insa o emisie permanenta de urina, picatura cu picatura, bloc cal, sau un sfincter anal hipoton.
Spina bifida chistica se asociaza in 90% din cazuri cu hidrocefalie, care este o complicatie majora a meningocelului, putand sa aiba aspect progresiv. Ea se poate evidentia de la nastere in 50-75% din cazuri.
O parte dintre copin cu spina bifida au deficienta mintala de grade diferite.
Tratamentul vizeaza repararea chirurgicala a defectului, cu incercarea de a inchide malformatia. Operatia este indicata in perioada neonatala, pana la maximum 3 luni. Pe langa interventia chirurgicala, tratamentul trebuie sa vizeze recuperarea neuromotorie si refacerea controlului sfincterian. Tratamentul preventiv este cea mai buna forma de terapie. Consta in administrarea acidului folic la femeile gravide si in special la cele care au avut un copil afectat anterior.
Prognosticul mielomeningocelului ramane rezervat, cel putin din punct de vedere al sanatatii.
Spina bifida oculta.
Termenul se refera la un disrafism spinal in care structurile nervoase sau meningele nu herniaza prin defectul mezenchimal, iar tegumentul suprajacent este normal. Acesta definitie include sinusul dermal, lipomul spinal, sindromul de notocord divizat (diplomielia), diastematomielia. Defectul mezenchimal este situat de obicei la nivel L5 S1. Defectul osos izolat (spina bifida radiologica) nu trebuie confundat cu spina bifida oculta, primul fiind un disrafism osos izolat, frecvent intalnit in practica, fara importanta clinica, spre deosebire de primul care include si disrafia partilor moi.
Sinusul dermal este un tub dermal captusit de epiteliu care se indreapta de la suprafata pielii catre sistemul nervos central. Multe dintre ele se termina in dura. Altele strapung dura terminandu-se intr-un chist dermoid intradural. Localizarea preferentiala este in regiunea sacrata sau occipitala. Sinusul dermal poate sa comunice sau nu cu structurile nervoase, de acest lucru depinzand aspectul clinic si complicatiile infectioase. Tegumentul de deasupra sinusului dermal este frecvent modificat, fiind hiperpigmentat sau cu pilozitate accentuata. Sinusul dermal este frecvent confundat cu sinusul pilonidal care este o depresiune situata numai la nivelul pielii (superficial).
Lipomul spinal este o colectie incapsulata de grasime si tesut conjunctiv, frecvent asociata cu o tulburare a fuziunii structurilor osoase posterioare.
Sindromul de notocord divizat (diplopmielia) rezulta din duplicatia maduvei in timpul embriogenezei, mai mult sau mai putin intinsa (pana la 10 segmente). Cele doua maduve sunt identice, dand nastere fiecare la patru radacini. In absenta altor malformatii asociate (spina bifida, lipom), acesta malformatie este muta.
Diastematomielia se caracterizeaza prin divizarea longitudinala a maduvei printr-un pinten osos sau cartilaginos median. Acest pinten, care poate fi redus numai la un tract fibros, ia nastere din arcul vertebral posterior si se indreapta anterior, traversand maduva. Cele doua parti ale maduvei astfel separate nu sunt functional separate asa cum se intamola in diplomielie. Localizatea cea mai frecventa este toraco-lombara, asociindu-se frecvent cu spina bifida oculta si cu anomalii cutanate sau cu alte malformatii ca: siringomielie, mielomeningocel. Tabloul clinic consta in deficit motor la membrele inferioare, spasticitate, scolioza progresiva. Pintenul osos poate fi operat.
Alte anomalii legate de disrafismul spinal include: siringomielia, siringobulbia, agenezia sacrala. Aceste anomalii au mecanisme diferite si numai partial anmintesc de disrafii.
Siringomielia este definita ca o cavitate tubulara in interiorul maduvei spinarii, tapetata cu celule gliale si necomunicanta cu ventriculul IV. Patogenia este necunoscuta, unii autori considera aceasta cavitate ca rezultat al unei anomalii de dezvoltare, in timp ce altii o considera fiind secundara unui traumatism medular care realizeaza o mielopatie chistica ascendenta. Semnele clinice decurg din nivelul localizarii cavitatii in plina subsatnta cenusie. Apare disociatia siringomielica cu afectarea suspendata (la membrele superioare, trunchi) a sensibilitatii termo-algice, in timp ce sensibilitatea tactila epicritica este conservata. Se asociaza tulburari trofice si vasomotorii. Largirea cavitatii poate afecta tracturile piramidale, rezultand hiperreflexie, spasticitate la membrele inferioare. La copil apare frecvent scolioza care poate fi primul semn, revelator de siringomielie. Daca cavitatea se situeaza la nivelul bulbului, afectiunea se numeste siringobulbie, care, de cele mai multe ori este insotita de o anomalie a oaselor de la baza craniului. Semnele revelatoare la copil sunt variabile: atingere asimetrica a uneia sau mai multor perechi craniene, pierderea disociata a sensibilitatii in teritoriul trigemenului, hemiatrofie si paralizie linguala, nistagmus. Atingerea vagului provoaca un stridor episodic, atingerea nucleului ambiguu un stridor cronic si o paralizie a corzilor vocale. Evolutia este foarte lenta, ramanand stabila ani de zile, pentru ca la un moment dat sa progreseze brusc si ireversibil.
Hidromielia reprezinta o dilatatie a canalului medular, captusita cu ependim, comunicanta cu ventriculul al IV-lea, care ar rezulta dintr-un obstacol in circulatia LCR, care ar antrena o hipertensiune intracraniana" class="text">hipertensiune intracraniana si o crestere a presiunii LCR in canalul medular cu dilatarea consecutiva a cavitatii (teoria hidrodinamica). Distinctia intre siringomielie si hidromielie este dificila in practica si se prefera ca pentru orice cavitate intraspinala de natura netumorala sa se vorbeasca de hidro-siringomielie.
Ambele sunt frecvent asociate cu malformatia Arnold Chiari.
Diagnosticul este confirmal de investigatiile radiologice. Radiografia simpla de coloana cervicala arata largirea canalului spinal, fie anomalii ale charnierei. RMN vizualizeaza cavitatea si ajuta la stabilirea indicatiei chirurgicale (chist sub tensiune, hiperpulsatil, necomunicant). Desi se poate practica decomprimarea fosei posterioare, in general se prefera tratament consevator.
Agenezia de sacru si coccis este o anomalie ososasa care se asociaza frecvent cu celelalte anomalii ale maduvei spinarii. Clinic se manifesta prin paraplegie, hipoplazie musculara, artrogripoza si ca neurogena flasca. Frecvent se produc infectii urinare recurente, rezistente la tratament.
II. Tulburari ale inductiei ventrale
Termenul desemneaza malformatii care rezulta din absenta diviziunii culelor telencefalice. Ele cuprind holoprozencefalia (arinencefalia). Acesta malformatie se produce cronologic in a 2-a luna de gestatie, perioada in care, dupa inchiderea tubului neural, survin trei evenimente importante: formarea culelor cerebrale (cele doua emisfere), evaginarea celor doua cule optice care vor forma tractul optic, evaginarea celor doua cule olfactive care vor forma tijele si bulbii olfactivi.
II.1. Holoprozencefalia si arinencefalia. Anomalia principala in holoprozencefalie este absenta diviziunii culelor telencefalice care determina formarea unei emisfere unice. Creierul are atunci forma unei hemisfere deschise in spate, asemanator unei potcoave. Cand se sociaza absenta procesului olfactiv, malformatia se numeste arinencefalie. Se asociaza de asemeni absenta diferentierii placii comisurale, astfel incat, in forma completa a malformatiei, nu exista nici corp calos, nici trigon, nici sept. Clinic se caracterizeaza prin anomalii ale fetei care in forma majora are aspect de ciclop, cu o fosa orbitara mediana unica, nasul este anormal sau absent sau redus la o narina unica. In formele mai usoare exista doar un hipertelorism extrem. Se poate asocia cheilo-gnato-palatoschizis. Exista retard mental sever si semne endocrine (nanism hipofizar, diabet insipid). Diagnosticul se pune pe baza aspectului CT cerebral.
III. Tulburari ale dezvoltarii cortexului sunt tulburari foarte frecvente ale tulburarii creierului. Frecventa lor a crescut in ultimul timp datorita explorarilor moderne (RMN). Ele sunt cauze de epilepsie, paralizii cerebrale, retard mental.
III.1. Tulburari de proliferare/diferentiere
Macrocefalia este cresterea perimetrului cranian cu mai mult de 2DS (deviatii standard) fata de valorile medii normale pentru varsta si sex; se poate datora mai multor cauze (Tabelul: Cauze de macrocefalie).
Macrocefalia se poate insoti de macroencefalie (creier de greutate, dimensiuni mai mari decat cele medii normale), de microencefalie (creier de dimensiuni reduse si greutate mica), rareori de un creier normal. Dintre cele enumerate in tabel, cauzele cele mai frecvente de macrocefalie sunt: hidrocefalia, macroencefalia, revarsatele lichidiene subdurale, edemul cerebral, displazia osoasa (cu ingrosarea calotei craniene). Exista macrocranie familiala, in care dezvoltarea intelectuala este normala si in care unul dintre parinti este macrocefal (se pare ca exista o transmitere autosomal dominanta). In celelalte forme de obicei se asociaza retard mental, sindrom piramidal si epilepsie secundara.
Microcefalia este scaderea perimetrului cranian cu mai mult de 2DS decat valorile medii normale stabilite pentru varsta si sex. Microcefalia este insotita obligatoriu de microencefalie existand posibilitatea afectarii neurologice si asociere cu deficit intelectual. Cand se pune diagnosticul de microcefalie, copilul trebuie investigat pentru depistarea cauzei acesteia (Tabelul: Cauze frecvente de
Tabelul: Cauze de macrocefalie (dupa Swaiman, modificat, 1994)
|
Grupa de cauze |
Exemple |
|
|
1. Hidrocefalie |
Comunicanta |
- bloc extraventricular (postinfectios, posthemoragic, insamantari maligne pe meninge |
|
Necomunicanta |
- malformatie Chiari - stenoza apeductala - malformatie Dandy-Walker - sindrom Walker-Warburg - neoplasme supra si infratentoriale - holoprozencefalie |
|
|
1. Macroencefalie (Tabelul: Cauze de macroencefalie) |
- anatomica - metabolica - hidrodinamica |
|
|
2. Colectie subdurala
|
- hematom - higroma - empiem |
|
|
3. Edem cerebral
|
- intoxicatii (plumb, vitamina A, tetracicline) - endocrin (hipoparatiroidism, hipoadrenocorticism) - galactozemie - degenerare spongiforma a creierului - psudotumor cerebri sindrom de HIC benign) |
|
|
4. Craniu ingrosat |
- variatii familiale - anemie - miotonie atrofica - displazii cranio-scheletale |
|
microcefalie si microencefalie). De asemenea trebuie diferentiata microcefalia, in care suturile pot fi inchise prematur, de craniostenoza primara a tuturor suturilor craniene, aceasta din urma beneficiind de tratament chirurgical care duce la dezvoltarea neuropsihomotorie normala a copilului.
Unii autori considera ca microcefalia vera este numai cea legata de defectul de proliferare. La celelalte cauze enumerate in tabel intervin si distructii celulare antenatale (microencefalia spuria).
Diagnosticul se pune cu ajutorul mijoacelor paraclinice imagistice (radiografia de craniu, ecografia transfontanelara, CT cerebral).
Tabelul: Cauze de macroencefalie (dupa Swaiman, modificat, 1994)
|
Grupa de cauze |
Exemple |
|
1. Macroencefalie anatomica |
- Focala (malformatie Oekonomakis, Lehrmitte-Duclos) - Unilaterala (fara hemihipertrofie somatica; cu hemihipertrofie somatica si hemangiomatoza/ hamartoame; sd. Klippel-Trenauny-Weber). - Bilaterala (asimptomatica familiala; simptomatica familiala; idiopatica simptomatica, nefamiliala) - Cu gigantism (sd. Sotos, de cauza hipofizara, adiposogigantism, sd. Weaver-Smith) - Cu hipostaturalitate (acondroplazie; hipostaturalitate tanatoforica; sd.FG; sd.Robinow; endocrinopatii multiple) - Cu sindroame neurocutanate ( neurofibromatoza; scleroza tuberoasa; hipomelanoza Ito; carcinom bazocelular nevoid; ataxia-telangiectazia) - Cu sindrom Klinefelter - Cu hemangioame, lipoame, hamartoame multiple - Diverse (distrofie musculara Duchenne; sd. Beckwith-Wiedemann; fibroelastoza endocardica si micropenie, etc.) |
|
2. Macroencefalie metabolica |
Boli lizozomale (gangliozidoza generalizata, mucopolizaharidoze, leucodistrofie metacromatica, boala Tay-Sachs) Degenerare spongiforma Canavan Boala Alexander Aminoacidurii/acidurii organice (boala cu urini cu miros de sirop de artar, deficienta de metilglutaril- CoA liaza) |
|
3. Macroencefalie hidrodinamica |
Scaderea drenajului venos sau al LCR |
|
4. Macroencefalie cu hidrocefalie |
- acondroplazie -
|
Hemimegalencefalia este hipertrofia unilaterala difuza a creierului. Morfologia generala a creierului este pastrata, girusurile sunt largite, iar consistenta cerebrala este ferma. Anatomo-patologic se descrie cresterea grosimii cortexului cerebral, cu prezenta unui mare numar de neuroni giganti hipercromatici. Clinic, de la nastere se observa o crestere in volum asimetrica a craniului.
III.2. Tulburarile de migrare implica tulburarile de deplasare a celulelor nervoase din zona germinativa periventriculara, deplasare care se realizeaza pe travee gliale pana la scoarta cerebrala. Plasarea celulelor nervoase in diferite straturi ale cortexului este determinata genetic.
Heterotopiile. Tulburarile care survin in perioada de migrare determina heterotopiile. Acestea constituie oprirea unor grupuri de celule nervoase in substanta alba in diferite locuri, in drumul lor spre scoarta. Mecanismul heterotopiilor este inca insuficient cunoscut. Se crede ca heterotopiile ar putea fi cauzate de o tulburare in mecanismul de moarte celulara (apoptoza), programat genetic. Se crede ca heterotopiile sunt implicate in etiopatogenia epilepsiilor. Diagnosticul lor se poate pune numai prin examinare prin rezonanta magnetica nucleara, CT cerebral avand un aspect normal.
Agirie-pahigirie (lisencefalie tip I, tip II). Termenul se refera la un creier cu o suprafata cerebrala neteda pe care este vizibila numai depresiunea vaii sylviene largite si putin adanci. Meningele este subtire si hipervascularizat. In cele mai multe cazuri exista cateva santuri alaturi de zone de polimicrogirie si pahigirie. Se descriu mai multe grade de lisencefalie depinzand de numarul santurilor, vizibile la RMN. Dupa numarul acestora, malformatia este denumita lisencefalie (fara girusuri) sau pahigirie (cu girusuri mari si putine). Din punct de vedere histologic, lisencefalia se imparte in tipul I si tipul II. In tipul I, cunoscut drept tipul clasic (Bielschowski), cortexul este foarte gros si este constituit din patru straturi. Neuronii din aceste straturi sunt anormal orientati, cu dendrite apicale, anormal orientate. Este caracteristica ectopia nucleilor olivari bulbari. Se poate asocia agenezia de corp calos. Cerebelul este de obicei normal. Cauzele lisencefaliei tipul I sunt multiple, majoritatea cazurilor fiind sporadice. Un numar important de cazuri au la baza o anomalie cromozomiala (deletia partii distale a bratului scurt a cromozomului 17 (17p 13.3). Astfel de cazuri fac parte din sindromul Miller-Dieker caracterizat clinic prin: frunte ingusta, filtrum lung, narine ridicate in sus, retrognatie, anomalii digitale si hipervascularizatia retinei.
Tipul II de lisencefalie (sindromul Walker-Warburg) este complet diferit de cel precedent din punct de vedere etiologic si morfologic. Si acest tip de
Tabelul: Cauze frecvente de microcefalie si microencefalie (dupa Swaiman, 1994)
|
Grupa de cauze |
Exemple |
|
|
1. Infectii |
- Toxoplasmoza - Rubeola - Citomegaloviroza - herpes simplex - sifilis - SIDA - Varicela - viroza cu Coxsackie B |
|
|
2. Medicamente/toxice |
- Citostatice - Medicamente antiepileptice - Alcool - Tutun - droguri (marijuana, cocana, heroina) - intoxicatie cu CO |
|
|
3. Hipoxie/ischemie |
- Patologie placentara (insuficienta) - status epilepticus - anoxie cerebrala generalizata |
|
|
4. Genetice
|
Cromo-zomiale |
- Sindrom Down si alte trisomii - Cromozomi inelari - aneuploidia cromozomilor sexuali - deletii |
|
Ereditare |
- familial (asimptomatic), cu intelect normal - simptomatic (dominant, recesiv, X-linkat) - craniosinostoze - boli degenerative heredofamiliale (Pelizaeus-Merzbacher, ceroid lipofuscinoza, aminoacidurii, sindrom Smith-Lemli-Opitz, sindrom de Lange, sindrom Rubinstein Taybi, sindrom Dubowitz, sindroame cu microstaturalitate armonica) |
|
|
5. Malformatii SNC |
- Microcefalie vera - holoprozencefalie - lisencefalie - hidranencefalie/porencefalie - encefalocel |
|
|
6. Afectiuni materne prenatale |
- anemie (Hb<10g/dl) - afectiuni cronice (pulmonare, infectii inclusiv TBC matern, cardiopatii) - convulsii (epilepsie, eclampsie) - malnutritie - expunere la toxice |
|
|
7. Traumatisme |
- traumatismele abdominale ale gravidei - nastere traumatica - copil batut |
|
|
8. Tulburari metabolice/endocrine perinatale |
- hipoglicemie - hipotiroidism - hipopituitarism - hipoadrenocorticism |
|
|
9. Malnutritie |
|
|
lisencefalie se transmite genetic, autosomal recesiv. Cortexul nu are santuri desi uneori pot exista zone de microgirie. Meningele este gros si are un aspect laptos datorat unei proliferari mezenchimale in special in jurul trunchiului cerebral. Cerebelul este mic, vermisul lipseste. Tractul piramidal este absent . Frecvent se asociaza hidrocefalie. Din punct de vedere microscopic, arhitectura corticala este complet anarhica, straturile coticale sunt subtiri si orientarea celulara este anormala, in diferite sensuri.
Clinic, in ambele tipuri exista intotdeauna encefalopatie infantila severa cu hipotonie axiala majora. Evolutia este in general fatala in primele luni, in special tipul II.
Diagnosticul lisencefaliilor este posibil cu ajutorul tehnicilor moderne de neuroimagerie (CT cerebral, RMN). EEG este sugestiv, traseele aratand o activitate alfa sau beta frecventa, de mare amplitudine.
Polimicrogiria. Termenul se refera la giri corticali anormali, ingusti, aglomerati, dand nastere unui model circumvolutional anormal. Polimicrogiria poate afecta intregul cortex, dar mai frecvent se localizeaza in anumite zone, in special in teritoriul arterei cerebrale mijlocii. Polimicrogiria se asociaza frecvent cu heterotopii sau cu agenezie de corp calos. Aspectul clinic este nespecific si depinde de extinderea, localizarea polimicrogiriei si de prezenta anomaliilor asociate.
III.3. Tulburari ale organizarii corticale
Displaziile corticale focale reprezinta modificari pe zone corticale limitate cu aspect macro- si microscopic anormal (arhitectura neuronala dezorganizata cu neuroni giganti si glie bizara). Din punct de vedere clinic tabloul este dominat de epilepsii focale si semne de deficit neurologic.
Microdisgeneziile corticale se refera la anomaliile corticale disgenetice minore.
IV. Tulburari ale diferentierii structurilor liniei mediane. Tulburarile placii comisurale sunt cele mai importante. Placa comisurala este o ingrosare a lamei terminale situata la extremitatea anterioara a tubului neural, la zona de jonctiune a celor doua cule telencefalice. Fibrele transversale ale placii comisurale, care constituie prima schita de corp calos, se dezvolta incepand cu saptamana 11-l2 de gestatie. Cresterea se face in acelasi timp spre inainte si spre inapoi, iar structura adulta se realizeaza spre a 18-20-a saptamana. Placa comisurala este o zona foarte vulnerabila la agresiuni (tractiuni provocate de o crestere rapida). Intre tulburarile de diferentiere a placii comisurale sunt incluse: agenezia de corp calos, schizencefalia si agenezia septo-optica.
Agenezia de corp calos (ura: Agenezia de corp calos) constituie cea mai importanta tulburare in diferentierea placii comisurale. Ea poate fi totala si partiala (anterioara sau posterioara). In forma completa, agenezia de corp calos poate asocia absenta comisurii anterioare si absenta comisurii hipocampice, caz in care nu exista nici o fibra transversala. Se pot asocia cu alte malformatii (tulburari de migrare). Etiologia este multipla, fiind implicati factori genetici (transmitere autosomal recesiva, X-linkata, autosomal dominanta, defecte cromozomiale trisomia 18, 13, 8), factori de mediu (sindromul de alcoolism fetal) si factori toxici endogeni (acidoza lactica, hiperglicinemia si alte defecte metabolice). De multe ori originea este necunoscuta.
Manifestarile clinice pot fi non-sindromice si sindromice. Formele non-sindromice sunt cele mai cunoscute. Ele pot fi asimptomatice sau evidentiate din cauza unei macrocranii. Copin pot avea retard mintal, crize epileptice si perimetru cranian crescut. Frecvent se evidentiaza hipertelorismsi, mai rar, anomalii endocrinologice. Formele sindromice include mai multe sindroame. Le enumeram pe cele mai importante:
- sindromul Aicardi asociaza spasme infantile sau/si crize partiale, lacune coroido-retiniene cu margini nete, coloboma al discului optic, anomalii costo-vertebrale, retard mintal sever. Microcrania apare in timp prin lipsa cresterii capului. Sindromul se intalneste numai la fete deoarece transmiterea este X-linkata, letala pentru baieti. Evolutia este severa, cu persistenta crizelor.
- Sindromul Shapiro se caracterizeaza prin agenezie de corp calos, hipotermie si episoade de hiperhidroza cauzate de leziunile hipotalamice.
- Sindromul Andermann asociala agenezie de corp calos si neuropatie periferica senzitivo-motorie transmisa autosomal recesiv (descrisa la canadienii frencezi).
- Sindromul oro-digito-facial (Papillon-Leage-Psaume) se caracterizeaza, pa langa agenezia de corp calos, care este inconstanta, printr-o hipertrofie a frenului lingual, gingiilor si limbii si prin polidactilie. Este transmisa X-linkat dominant, letala pentru baieti.
- Lipomul de corp calos se asociaza cu agenezie partiala a acestei comisuri.

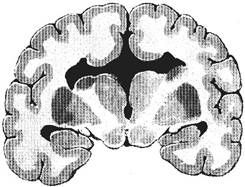
ura: Agenezia de corp calos
Agenezie septala (displazia septo-optica - sindromul De Morsier). La agenezia septala se asociaza o hipoplazie a tractului optic. Sindromul se caracterizeaza prin hipopituitarism. Cinic nou-nascutul prezinta hipotonie si hipoglicemie precoce si severa. Papila nervului optic are un aspect de dublu contur cu o coroana pigmentara in jur.
V. Malformatiile cerebelului si ale fosei posterioare
Disgeneziile cerebelului pot fi izolate sau asociate cu alte anomalii de dezvoltare ale SNC.
Malformatia Dandy-Walker cuprinde agenezia completa sau partiala a vermisului, o formatiune chistica de mari dimensiuni in fosa posterioara care corespunde cu o expansiune diverticulara a ventriculului IV, enorm dilatat, si hidrocefalie care lipseste la nou-nascut si la sugar si uneori chiar pana tarziu in viata adulta. Se asociaza frecvent tulburari de migrare neuronala, agenezie de corp calos, encefalocel occipital, cheilo-palatoschizis, malformatii cardiace, ale tractului urinar si dismorfism facial. Uneori apare atrezia gaurii Magendie si Luschka. Sindromul este atribuit unei opriri in dezvoltarea mezencefalului, cu persistenta membranelor ventriculului IV fetal. Manifestarile clinice sunt in principal cele ale hidrocefaliei. De remarcat ca nu apar semne de disfunctie cerebeloasa. Frecvent se asociaza retard mintal. Tratamentul implica operatie de drenare a hidrocefaliei si nu deschiderea cavitatii chistice (ventricului IV).
Agenezia de vermis se refera la defectul median in care vermisul este inlocuit de o membrana translucida. Poate fi completa sau partiala (in special partea posterioara). Se poate insoti uneori de fuziunea emisferelor cerebeloase pe linia mediana. Clinic se caracterizeaza prin sindromul dezechilibrului (ataxie statica si retard in achizitia posturii) descris de Hagberg.
Sindromul Joubert consta in agenezia familiala a vermisului cerebelos, episoade de hiperpnee, miscari oculare anormale, ataxie si retard. Se pot asocia asimetrii faciale, anomalii retiniene.
Agenezia emisferelor cerebeloase este mai rara. Poate fi uni- sau bilaterala. Poate sa respecte vermisul si lobul floculonodular. Aplazia cerebeloasa totala este exceptionala si nu este totdeauna asociata cu manifestari clinice evidente. Mai frecvent se intalneste o hipoplazie unilaterala.
VI. Chisturi intracraniene sunt procese expansive cu continut lichidian, netumorale si neinfectioase. Chisturile arahnoidiene se dezvolta intre creier si baza creierului sau convexitate. Peretele lor este adesea arahnoidia. Cel mai frecvent sunt situate intre arahnoida si pia mater. De obicei sunt asimptomatice sau se evidentiaza la nastere printr-o macrocranie sau mai tarziu printr-o complicatie. Localizarea cea mai frecventa este supratentorial in valea sylviana. Pot fi relevate printr-o hemoragie subdurala dupa traumatisme cerebrale minore. In general nu necesita tratament.
VII. Malformatiile charnierei occipitocervicale
Malformatia Arnold Chiari consta in anomalii de pozitionare a jonctiunii bulbo-medulare si a cerebelului care are tendinta de a hernia in gaura occipitala (ura: Malformatia Arnold Chiari). Exista mai multe tipuri de malformatie, dintre care doua sunt mai importante.
Tipul I consta in pozitia anormala a cerebelului care este deplasat caudal, spre gaura occipitala, ajungand uneori pana la C3. Se poate asocia hernierea amigdalelor cerebeloase, fibroza meningeala si siringomielia maduvei cervicale sau alte anomalii ale bazei creierului (platibazie) sau a vertebrelor cervicale (sindromul Klippel-Feil). Malformatia Chiari I devine simptomatica de obicei la adolescenta, cu cefalee, dureri cervicale, semne motorii si senditive, paralizii de nervi cranieni, semne cerebeloase. La copil este cel mai frecvent asimptomatica.
Tipul II asociaza la semnele anterior descrise mielo-meningocel si hidrocefalie.
Sindromul Klippel-Feil este o malformatie sporadica constand in fuziunea vertebrelor cervicale care apare devreme in viata intrauterina (saptamana a6-a). Clinic apare un gat scurt si o imtatie joasa a parului. Poate sa fie asimptomatica sau pot exista semne neurologice care se pot agrava brusc la un traumatism minim (semne de compresiune medulara).

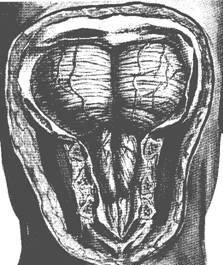
ura: Malformatia Arnold Chiari
MALFORMATII TARDIVE
Cuprind porencefalia si schizencefalia.
Porencefalia reprezinta o cavitate chistica (porus) circumscrisa care se formeaza prin distructia cerebrala focala urmata de resorbtie tisulara (ura: Porencefalia). Cavitatea este de obicei localizata in teritoriul sylvian. Uneori este redusa la o simpla fanta ingusta, orientata spre ventricul cu care de obicei comunica. Poate sa fie uni- sau bilaterala. Marginile porencefalie sunt de obicei formate dintr-un cortex cu microgirie. Aspectul clinic al porencefaliei variaza dupa localizarea si intinderea ei. Manifestarile clinice importante sunt: intarzierea mintala, epilepsia, semne neurologice de deficit focal (hemiplegie, tetraplegie, hemianopsie). Uneori porencefalia creste progresiv in volum antrenand o deformare a creierului si putandu-se acompania cu o agravare clinica.


ura: Porencefalie
Schizencefalia (hidranencefalia) este considerata ca o forma extrema de porencefalie. Distrugerea se intinde in acest caz la cea mai mare parte a emisferelor. Topografia distrugerii corespunde teritoriului de irigare a carotidelor interne. Cortexul distrus este inlocuit cu o membrana gliala.
Sistemul nervos vegelaliv (SNV) inerveaza musculatura neleda a vaselor si viscerelor, glandele exocrine si endocrine si anumite celule ale tesuturilor [...] |
ingrijirea pacientilor cu tumori primare sau metastatice ale SNC necesita: (I) stabilirea cu acuratete a diagnosticului de tumora si excluderea altor [...] |
Sistemul nervos vegelaliv (SNV) inerveaza musculatura neleda a vaselor si viscerelor, glandele exocrine si endocrine si anumite celule ale tesuturilor [...] |
Copyright © 2010 - 2025
: eSanatos.com - Reproducerea, chiar si partiala, a materialelor de pe acest site este interzisa!
Informatiile medicale au scop informativ si educational. Ele nu pot inlocui consultul medicului si nici diagnosticul stabilit in urma investigatiilor si analizelor medicale la un medic specialist.
Termeni si conditii - Confidentialitatea datelor - Contact