|
|
Hemofilia A
Hemofilia A este o tulburare a coagularii transmisa legat de X, determinata de o deficienta a factorului VIII plasmatic al coagularii. Factorul VIII al coagularii are rolul de cofactor al factorului IX, o proteaza care regleaza activarea factorului X.
Incidenta. Hemofilia A are o incidenta de aproximativ 1 la 5.000-l0.000 de nou-nascuti de sex masculin.
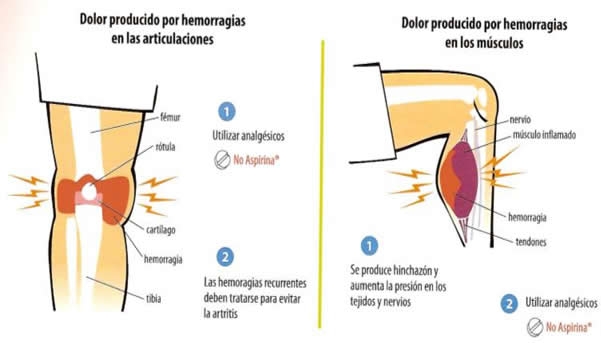
Simptomatologie.
Manifestarile
clinice sunt prezente de regula la indivizi de sex masculin, desi
exista si cazuri rare in care apar manifestari clinice la femei
care au inactivare preferentiala a cromozomului X, fara
mutatie. Boala este caracterizata prin hemoragii la
traumatisme minime care pot persista pe durata catorva ore sau zile si
care conduc la formarea de hematoame la nivelul tesuturilor moi si al
muschilor, precum si a hemoragiilor intraarticulare, care
evolueaza spre artroza. Formele severe de boala sunt
diagnosticate in perioada neonatala datorita prezentei unor
hematoame cefalice excesive sau a hemoragiei prelungite la nivelul cordonului
ombilical. Formele moderate dezvolta hematoame si hemartroze o
data cu inceputul mersului, in timp ce in formele minore diagnosticul
poate fi pus cu ocazia unor interventii chirurgicale sau a unor
traumatisme.
Diagnostic. Diagnosticul se bazeaza in mod curent pe masurarea activitatii factorului VIII al coagularii. Diferentierea trebuie facuta in primul rand de hemofilia B, determinata de mutatii ale genei pentru factorul IX al coagularii. Aceasta boala este identica din punct de vedere clinic, dar incidenta este mai redusa (1 la 100.000). In functie de nivelul activitatii factorului VIII, hemofilia A poate fi clasificata in forma usoara (activitate restanta de 5-25%), forma moderata (activitate restanta de 1-5%) si forma severa (activitate restanta sub 1%).
Genetica. Hemofilia A este determinata de mutatii ale genei F8C, localizata la nivelul cromozomului Xq28. Mutatiile sunt extrem de variate si includ deletii, insertii, inversii si mutatii punctiforme. Cea mai frecventa mutatie - care este responsabila de aproximativ 25% dintre cazurile de boala si circa 40-50% dintre formele severe - este o inversie ce asociaza si o deletie a portiunii carboxiterminale a factorului VIII. O alta categorie de mutatii asociate cu forme severe de boala sunt insertiile unor elemente retrotranspozabile L1 in interiorul genei. Dintre mutatiile punctiforme, cele asociate unor forme severe sau moderate de boala sunt cele care determina aparitia unor codoni stop prematuri, cele care altereaza capacitatea de legare a factorului VIII la factorul von Willebrand sau la factorul IX al coagularii ori cele care afecteaza capacitatea proteinei de a fi secretata.
Tratamentul standard actual consta in inlocuirea intravenoasa a factorului VIII. Acest tratament a permis prelungirea sperantei de viata de la sub 2 ani la inceputul secolului XX la circa 65 de ani in prezent.
Sfat genetic. Daca o femeie are istoric familial de hemofilie, statusul sau purtator poate fi determinat prin analize de inlantuire sau prin screening-ul mutatiei genei F8C atunci cand aceasta a fost identificata deja la o ruda. Exista si teste care pot fi utilizate pentru analiza prezentei celei mai frecvente mutatii, inversia mai sus amintita. Determinarea statusului de purtator prin metode enzimatice este dificila. Daca femeia este purtatoare, ea are un risc de 50% de a da nastere unor baieti bolnavi si un risc egal de a da nastere unor fete purtatoare. Avand in vedere frecventa inactivarii preferentiale a cromozomului X exista si un risc mic de a se naste o fata cu simptome de hemofilie. In cazurile in care mama are un baiat cu hemofilie, dar nu exista istoric familial pozitiv, trebuie luata in calcul posibilitatea unor mutatii de novo, care sunt responsabile pentru circa 1/3 dintre cazurile de boala. In acest caz, riscul de recurenta a bolii este in general mai mic.
Bolile genetice ale hemoglobinei - prototip de boli moleculare
Bolile genetice ale hemoglobinei (Hb) se impart in trei mari categorii patogenice: variante structurale care sunt modificari in structura lanturilor de globina, fara afectarea ratei lor de sinteza; talasemiile, produse prin sinteza scazuta a unu sau mai multe lanturi de globina; persistenta ereditara a hemoglobinei fetale, determinata de o perturbare a comutarii' perinatale de la sinteza γ-globinei, la β-globina.
(1) Cele circa 400 de Hb anormale, produse de obicei prin mutatii punctiforme in genele globinelor, se subimpart in trei clase:
Variantele ce produc anemie hemolitica se clasifica in:
- Hemoglobine anormale ce capata noi proprietati. Hemoglobina S, din sicklemie ( l.p.), rezulta prin inlocuirea Glu6Val in catenele de β-globina; acest fapt nu modifica capacitatea de fixare a oxigenului, dar scade drastic solubilitatea Hb (in sangele dezoxigenat), care se polimerizaeaza - producand fenomenul de sicklizare; acesta genereaza hemoliza eritrocitelor si microtrombozele capilare. Hemoglobina C (substitutia Glu6Liz), in forma sa oxigenata, tinde sa cristalizeze, reducand deformabilitatea hematiilor si generand astfel o hemoliza medie;
- Hemoglobine instabile (de exemplu, Hb Gun Hill) sunt determinate de mutatii ce denatureaza tetramerul de Hb; ele devin insolubile si precipita, formand corpii Heinz, ce contribuie la producerea hemolizei.
Variante ce altereaza transportul de oxigen; in acest grup se includ:
- Methemoglobinele (de exemplu, Hb Hyde Park), care datorita oxidarii Fe2+ din hem la Fe3+ -rezistent la methemoglobinreductaza - sunt incapabile de oxigenare reversibila;
- Hb cu modificarea afinitatii pentru oxigen (de exemplu Hb Kempsey sau Kansas)
(2) Talasemiile sunt, in ansamblul lor, cele mai frecvente boli monogenice. Mutatiile altereaza sinteza sau stabilitatea lanturilor de α-globina sau de β-globina, producand alfa-talasemii sau beta-talasemii. Catenele ce se sintetizeaza normal vor fi in exces, pot precipita alterand membrana hematiilor si genera distrugerea lor. Frecventa talasemiilor este crescuta in bazinul Mediteranean si zonele tropicale, subtropicale din toata lumea, datorita avantajului selectiv al heterozigotilor fata de malarie.
Alfa-talasemiile se caracterizeaza prin absenta lanturilor de α-globina (deletii prin crossing-over inegal); Hb va fi formata numai din lanturi beta (tetramerul β4 sau Hb H) sau, in perioada fetalas numai din lanturi gama (teramerul γ4 sau Hb Bart); ambele sunt transportori ineficace de oxigen, generand hipoxie. Gravitatea bolii depinde de numarul de lanturi α deletate: αα/α-, purtator sanatos; α- /α-, trasatura α-talasemica (anemie usoara); HbH(β4), α-/-- , anemie hemolitica severa; Hb Bart (γ4), -- /--, ce produce hidrops fetalis letal.
Beta-talasemiile se caracterizeaza prin scaderea productiei lanturilor de β-globina; excesul lanturi de α-globina precipita generand anemie hemolitica. In locul lanturilor β se sintetizeaza fie lanturi δ si astfel creste cantitatea de Hb A2, fie lanturi γ si atunci creste sinteza HbF. Indivizii homozigoti, cu doua gene β mutante, prezinta o forma severa de anemie si boala se numeste talasemia major (cu diferite forme, functie de cantitatea de HbA ce se poate forma: β0-talasemia, cand absenta de sinteza a lanturilor β este aproape completa si nu se formeaza HbA; β+-talasemia cu Hb A detectabila). Indivizii heterozigoti, cu o singura alela β mutanta, au o usoara anemie microcitara hipocroma (talasemia minor sau trasatura beta-talasemica); detectia lor - prin cresterea cantitatii de HbA1 - este utila pentru sfatul genetic.
(3) Persistenta ereditara a Hb fetale este reprezentata de un grup de afectiuni, relativ benigne, caracterizate prin imposibilitatea comutarii postnatale a sintezei lanturilor γ de Hb la sinteza lanturilor β; in consecinta pacientii au un exces de HbF (α2γ2) si o scadere a productiei de HbA (α2β2).
Definitie: este o afectiune congenitala, transmisa ereditar X-linkat, caracterizata prin sinteza scazuta sau anormala a factorului IX. Hemofi [...] |
Boala polichistica renala autozomal dominanta Boala polichistica renala a adultului (ADPKD) este o boala multisistemica ereditara, p [...] |
Profilaxia bolilor genetice include un ansamblu de masuri ce urma [...] |
Copyright © 2010 - 2025
: eSanatos.com - Reproducerea, chiar si partiala, a materialelor de pe acest site este interzisa!
Informatiile medicale au scop informativ si educational. Ele nu pot inlocui consultul medicului si nici diagnosticul stabilit in urma investigatiilor si analizelor medicale la un medic specialist.
Termeni si conditii - Confidentialitatea datelor - Contact